Auschecken über Ihr Konto
Als Neukunde auschecken
Ein Konto zu erstellen hat viele Vorteile:
- Bestellungen und Sendungen verfolgen
- Alte Bestellungen einsehen
- Schneller zur Kasse gehen
Methodenentwicklung
Die Methodenentwicklung in der Chromatographie ist ein systematischer Prozess, um eine analytische Methode zu erstellen, die spezifisch, zuverlässig und effizient Probenkomponenten trennt. Dies umfasst die Auswahl der geeigneten Chromatographieart, die Bestimmung der optimalen Säulen- und Mobilphasenbedingungen, sowie die Einstellung der Detektorparameter. Ziel ist es, eine Methode zu entwickeln, die eine hohe Auflösung der Analyten bietet, reproduzierbare Ergebnisse liefert und den analytischen Anforderungen entspricht. Dieser Prozess erfordert tiefgehendes Wissen über die chemischen Eigenschaften der Analyten und die Interaktionen mit der stationären Phase.
Über MZ-Analysentechnik GmbH erhalten Sie Zugriff auf nahezu das gesamte Spektrum an weltweit erhältlichen stationären Phasen, damit Sie für Ihre Analyse stets die richtige Säule parat haben. Gern helfen wir Ihnen bei der Auswahl Ihrer passenden Säule.
Die Schritte einer typischen Methodenentwicklung könnten folgendermaßen aussehen:
- Auswahl der Trenntechnik (RP, NP, HILIC, SEC, IEX,...)
- Auswahl der Säulendimension
- Auswahl der stationären Phase
- Auswahl der mobilen Phase
- Einstellung des pH-Wertes der mobilen Phase mit einem geeigneten Pufferreagenz (falls benötigt)
- Durchführung einiger isokratischen oder Gradientenläufe um grundlegende Parameter festzulegen
- Weitere Optimierung der Parameter, z.B. durch Variation
- der Temperatur
- des Puffer- oder Ionenpaarreagenz
- der Flussgeschwindigkeit
- des Gradienten
Einige Hersteller stellen zum Thema Methodenentwicklung hilfreiche Literatur bereit, die es dem Anwender erleichtern für sein Trennproblem die optimalen Bedingungen zu finden. Sehen Sie sich dazu bitte untenstehende Broschüren an. Bei Fragen zum Thema Methodenentwicklung oder der Säulenauswahl stehen wir Ihnen gern zur Verfügung.
Technische Informationen
Die chromatographische Methodenentwicklung verfolgt das Ziel eine bestimmte Anzahl von Analyten in möglichst kurzer Zeit und bestmöglicher Auflösung zu trennen. Gleichzeitig sollen die Methoden reproduzierbar und robust sein, damit zu unterschiedlichen Zeiten, an verschiedenen Orten identische Ergebnisse erhalten werden können.
Die Auflösung (Rs) einer Trennung wird durch die folgende Gleichung beschrieben:
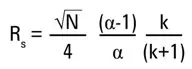
Die Auflösung wird demnach durch die Effizienz N, die Selektivität α und die Retention k beeinflusst.
Die Effizienz N einer Säule ist u.A. von deren Länge, der Partikelgröße und der Partikelform abhängig. Sie wird durch die theoretische Bodenzahl einer Säule beschrieben. Die Selektivität α wird hauptsächlich beeinflusst von der Art der stationären Phase und der mobilen Phase. Die Retention k kann ebenfalls durch die Art der stationären Phase und durch die Elutionskraft der mobilen Phase beeinflusst werden.
Als Beispiel: Bei Verdoppelung der Bodenzahl (entweder durch doppelte Säulenlänge oder Verkleinerung des Partikeldurchmessers) erhöht sich die Auflösung um den Faktor Wurzel 2, d.h. lediglich um etwa 40%. Dabei erhöht sich jedoch die Analysendauer und/oder der Rückdruck erheblich. Zu bedenken ist auch, dass bei Halbierung des Partikeldurchmessers sich der Rückdruck vervierfacht.
Aus der graphischen Auftragung der Auflösung gegen die Effizienz, Selektivität und Retention lässt sich erkennen, dass die Auflösung einer Trennung am stärksten von der Selektivität beeinflusst wird.
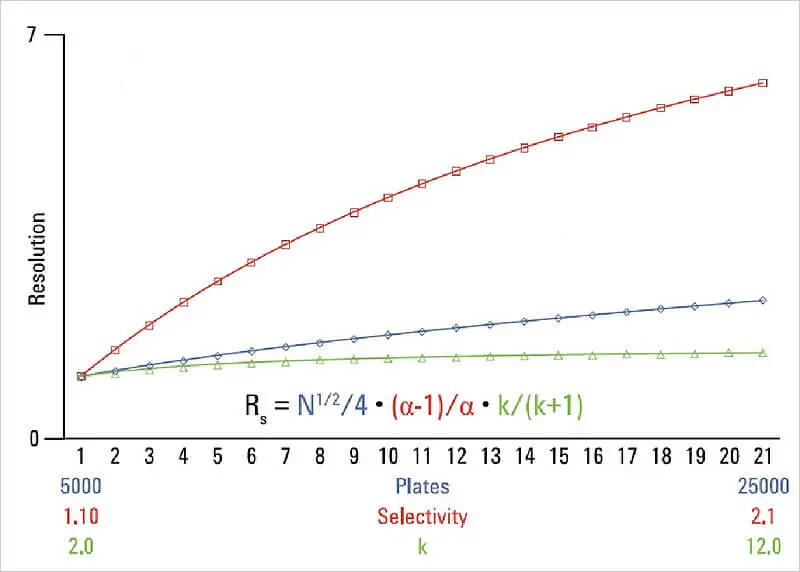
Aus diesem Grund ist die Selektivität der stationären Phase einer der nützlichsten Parameter zur Einstellung der HPLC-Trennungsselektivität und somit zur Optimierung der Auflösung. Die Methoden lassen sich vor allem zwischen den Labors leichter reproduzieren, wenn die Selektivität bei Verwendung einfacher mobiler Phasen aus der stationären Phase stammt. Hat die stationäre Phase die passende Selektivität für das gegebene Trennproblem, so lässt sich bei der darauffolgenden Optimierung einiges an Arbeit sparen.
Warum ist die Kontrolle des pH Wertes wichtig?
Ionisierung der Analyten
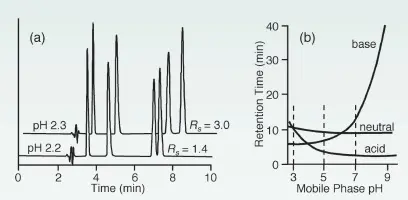
Ionisierbare Verbindungen (wie Säuren oder Basen) liegen bei verschiedenen pH Werten entweder ionisiert oder neutral vor. Unterscheidet sich der pH Wert um mehr als 2 Einheiten vom pka Wert einer Verbindung, dann ist diese zu 99% ionisiert oder neutral. Wenn diese Verbindung neutral vorliegt, dann ist diese auch hydrophober, was zu einer längeren Retentionszeit in der Umkehrphasenchromatographie führt. Bei Säuren ist das für pH < pka und für Basen bei pH > pka der Fall. Liegt der pH Wert nahe dem pka Wert einer Komponente, können bereits kleine Änderungen des pH Wertes zu großen Unterschieden im Grad der Ionisierung und somit auch der Retention führen.
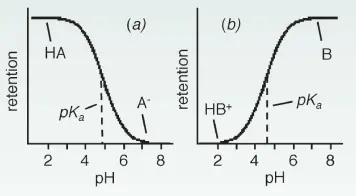
Ionisierung der stationären Phase
Neben den Analyten, beeinflusst der pH Wert auch die Säule. Zunächst einmal muss der pH Wert in den Grenzen der Säule liegen. Für Silica Gele sind diese meist 2<pH<8. Ein weiterer wichtiger Aspekt ist die Deprotonierung der freien Silanol Gruppen. Bei älteren “Typ A” Silica Gelen liegt der pka Wert dieser freien Silanole bei 4-5. Das bedeutet, dass sich in diesem pH Bereich die Ionisierung der stationären Phase stark ändern kann. Liegen die freien Silanole deprotoniert vor, kommt es durch die negativ geladenen Gruppen zum Kationenaustausch und einem starken Tailing für Basen. Daher werden heutzutage meist neuere “Typ B” Silica Gele eingesetzt, die einen pKa Wert von >7 aufweisen und in der Regel auch deutlich symmetrischere Peaks für Basen zeigen.
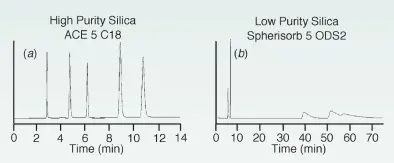
Beginn der Methodenentwicklung
Fazit: Auf Grund der Ionisierung von Analyten und der stationären Phase ist ein Start der Methodenentwicklung bei einem pH Wert von 2-3 empfohlen. Bei diesem pH Wert liegen die meisten organischen Säuren neutral und die Basen geladen vor. Gleichzeitig ist auch die stationäre Phase neutral und retardiert dabei die Säuren stärker als die Basen. Dies führt dazu, dass Basen zuerst eluieren. Sollten die pka Werte der Analyten bekannt sein, ist es außerdem sinnvoll einen pH Wert um zwei Einheiten entfernt von den pka Werten zu verwenden, um eine robuste Methode zu erhalten.
Welche Puffer können verwendet werden?
Ein Puffer ist am effektivsten im Bereich von ±1 pH um seinen pka Wert. Da in der Chromatographie aber nur wenig Probe injiziert wird kann ein pH Bereich von ±2 um den pka Wert ausreichend sein.
| Puffer | pka | pH Bereich | LC-MS kompatibel |
| Phosphat (pk1) | 2.15 | 1.1 - 3.1 | - |
| Phosphat (pk2) | 7.20 | 6.2 - 8.2 | - |
| Phosphat (pk3) | 12.3 | 11.3 - 13.3 | - |
| Acetat | 4.8 | 3.8 - 5.8 | + (als Ammoniumacetat) |
| Trifluoressigsäure (TFA) | 0.3 | 2.0 | + (aber unterdrückt Ionisierung) |
| Phosphorsäure | 2.15 | 2.0 | - |
| Ameisensäure | 3.75 | 2.7 | + |
| Ammoniumformiat | 3.7 | 2.7 - 4.7 | + |
| Ammoniumbicarbonat | 7.6 | 6.6 - 8.6 | + |
| Borat | 9.3 | 8.3 - 10.3 | - |
 Avantor ACE HPLC Buffer Solution Guide
Avantor ACE HPLC Buffer Solution Guide